注:如需PDF原文,请将E-mail发送至本公司邮箱,注明所需文章即可。
王介强, 陶珍东, 孙旭东
摘要: 以廉价无机盐为原料, 采用溶胶凝胶法制备出尺度均匀, 一次颗粒尺寸平均为60 nm , 颗粒呈球形的高纯Y2O3 纳米微粒。研究发现适量SO42 - 离子的添加对生成前驱物溶胶及煅烧得到球形的Y2O3 纳米微粒起关键作用。对前驱物在煅烧过程中的物相变化进行了研究, 分析了煅烧温度对产物粒度和纯度的影响, 结果表明在不使生成物颗粒过分长大的前提下, 升高煅烧温度有助于制取高纯和晶化完全的Y2O3 微粉。
关键词: 无机非金属材料; 纳米微粒; Y2O3 ; 无机溶胶凝胶; SO42 - ; 煅烧温度; 稀土
随着稀土应用研究的广泛和深入发展, Y2O3作为一种重要的稀土氧化物原料, 在新材料特别是功能材料研究方面, 其重要性日渐突现; 如以Y2O3 为主要原料制取的广泛用于医疗领域的激光晶体YAG, 用于通讯领域的微波磁性材料YIG, 彩色显像行业用的荧光材料Y2O3∶Eu , 优良光学特性的Y2O3 系透明陶瓷等, 此外Y2O3 还是ZrO2 陶瓷的常用稳定剂以及AlN , Si3N4 等高温结构陶瓷材料常用的烧结助剂。上述材料的制备对Y2O3 原料不仅要求高的纯度, 还要求良好的烧结活性。尽管工业上采用离子交换法或溶剂萃取法已生产出纯度可达99. 99 %或99. 999 %的Y2O3 粉料, 但工业制粉的烧结活性尚不能满足上述材料制备的使用要求, 因此开发具有高活性的Y2O3纳米微粒具有重要的意义。本文以工业高纯Y2O3粗粉为原料, 在不降低其纯度的前提下, 通过无机溶胶2凝胶法, 制取了具有高活性的Y2O3纳米微粒, 并对制取工艺、影响因素进行了分析和研究。
1 实验方法
将工业生产的纯度为99. 99 % , 平均粒径5μm的Y2O3粉溶于分析纯的浓硝酸, 加去离子水配制成浓度为0. 26 mol·L - 1的Y(NO3)3 溶液, 称取微量分析纯的(NH4)2SO4 添加到Y(NO3)3 溶液中, 用磁力搅拌器搅拌数分钟, 将浓度为1 mol·L - 1的氨水溶液滴加到剧烈搅拌的Y(NO3)3 溶液中, 滴定时将反应体系置于冰水混合物中, 使反应体系的温度维持在0 ℃上下, 氨水的滴加速度控制在2 ml·min -1以下, 当反应体系的pH = 8. 5 时, 停止滴加氨水, 继续搅拌溶液, 时效5 h , 时效温度仍维持在0 ℃上下, 时效结束后, 得到前驱物溶胶,将溶胶用去离子水洗涤两次, 以去除溶胶中多余的NO3- , NH4+ 等离子, 再用丙酮将溶胶洗涤两遍, 以去除溶胶中多余的水份, 而获得前驱物凝胶, 凝胶干燥采用室温下氮气吹干。将干燥后的前驱物粉料装入石英烧舟内, 于流动的氧气气氛中在不同温度下进行煅烧。
采用日本岛津DT30 型DTA 分析仪研究前驱物在加热过程中的物相变化, 升温速度为10 ℃·min -1 , 气氛为流动的氧气。采用日本理学D/MAX2RB 型X 射线衍射仪分析前驱物和其在不同煅烧条件下产物的物相组成。采用PhilipsEM420型透射电子显微镜(TEM) 观测前驱物及其在不同煅烧条件下产物的一次颗粒尺寸和形貌。
2 结果与分析
2. 1 SO42 - 对前驱物和煅烧产物粒度和颗粒形貌的影响
为研究SO42 - 对前驱物和煅烧产物粒度和颗粒形貌的影响, 在其他条件相同的情况下, 采取向前驱物生成体系中不加(NH4 )2SO4 和加入微量(NH4) 2 SO4 以进行对比。结果发现没有(NH4)2SO4加入时, 反应生成的前驱物将很快沉淀下来, 而有适量(NH4) 2 SO4 加入时, 则可得到前驱物溶胶, 图1 和2 分别为两种情况下所得的前驱物颗粒及其在1100 ℃下保温3 h 所得Y2O3 颗粒的TEM 形貌。

由图1 可以看出, 没有加入(NH4)2SO4的反应体系, 由于严格控制了滴加工艺, 生成的前驱物其一次颗粒的尺寸和形貌较为理想, 但一次颗粒之间存在明显团聚, 这是导致前驱物很快沉淀下来的主要原因, 但这种团聚在室温以下尚处于一种软团聚的形式。而煅烧所得的Y2O3 颗粒尺寸在500 nm~1μm 不等, 并且粒形极不规则, 表明煅烧所得的Y2O3 颗粒已不能保持其前驱沉淀物的颗粒形貌, 在煅烧过程中, 前驱物颗粒间的软团聚发展为硬团聚, 并且具有各向异性, 导致长大后的颗粒尺寸不均, 形状也不规则。
从图1 和2 还可看出, 当反应体系添加适量(NH4)2SO4时, 得到的前驱物溶胶颗粒呈雪花状的分散体, 前驱物颗粒基本呈线状排列的形式, 这种开放式的结构体系使得前驱物颗粒不易发生团聚而沉淀, 1100 ℃煅烧后, 得到尺寸均匀的Y2O3球形微粒, 平均尺寸为60 nm。表明在煅烧过程中呈线状排列的前驱物颗粒发生分解, 分离, 生成的Y2O3 在晶化过程中呈各向同性发展。这表明SO42- 具有影响前驱物颗粒排列方式、抑制前驱物颗粒团聚的作用, 在煅烧过程中还起到促进颗粒晶化各向同性发展的作用。其他学者也发现SO42- 对液相法制取纳米微粒有类似的影响, 但对SO42- 的加入量应严格控制,添加过多的SO42- 会由于SO42- 与阳离子较强的亲合力, 在前驱物洗涤和煅烧过程中不易脱除, 因此影响Y2O3 微粉的纯度,起到相反的效果。
上述结果表明, 由于SO42- 的影响使得前驱物和其煅烧产物的颗粒聚集状态及其尺寸和形貌都发生了巨大差别。通过适量SO42- 离子的添加, 并加以对滴加工艺进行控制可获得前驱物溶胶并制备出高活性的Y2O3 纳米微粒, 这种以廉价无机盐为原料通过溶胶凝胶法制备纳米微粒的方法, 显然较采用昂贵的金属醇盐为原料的溶胶凝胶法更有前途。
2. 2 前驱物在煅烧过程中的物相变化及煅烧温度对煅烧产物的影响
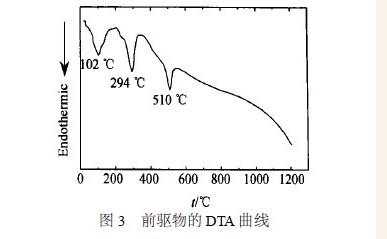
图3 为前驱物的DTA 曲线, 从图中可以看出,各峰均为吸热峰, 表明在该处发生了明显的物相变化, 结合图4 中前驱物及其在不同煅烧条件下产物的XRD 曲线可以看出: 前驱物的主要成分为Y2 (OH)5(NO3) 0. 86·H2O , 前驱物的DTA 曲线于102℃处的吸热峰主要是因前驱物中的吸附水、结晶水和结构水脱除引起的; 此时, 前驱物Y2 (OH)5(NO3) 0. 86·H2O 转变成α2Y(OH)3 相, 在294 ℃的吸热峰表明α2Y(OH)3 进一步失水转变为单斜结构的YOOH 相, 在510 ℃的吸热峰表明中间相开始向体心立方结构的Y2O3 相转变; 因此,前驱物在煅烧过程中的物相变化为: Y2(OH)5(NO3) 0. 86·H2O →α2Y(OH)3 →YOOH →Y2O3 。
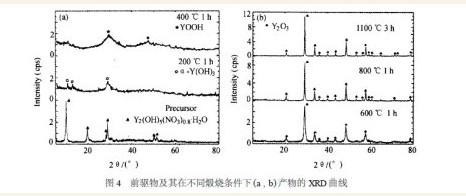
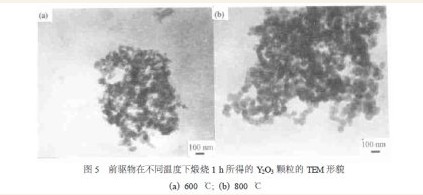
由图4 中前驱物在高于600 ℃温度下煅烧产物的XRD 曲线表明, 虽然600 ℃保温1 h 得到的煅烧产物已主要为Y2O3 , 但对比其在800 和1100℃的煅烧产物Y2O3 的衍射峰加强说明, 在600 ℃以下, 相变反应及Y2O3 的结晶都不完全, 因此要得到晶化完全且高纯的Y2O3 微粉, 应该把煅烧温度升高到1100 ℃。根据谢乐公式, 随煅烧温度的升高, 煅烧产物Y2O3 的衍射峰变得尖锐, 表明其晶粒尺寸随温度升高而长大。图5 分别为前驱物在600 和800 ℃下保温1 h 得到的煅烧产物的颗粒形貌, 与图2 中在1100 ℃下保温3 h 得到的煅烧产物相比, 随煅烧温度的升高颗粒尺寸只是略有长大, 并不显著, 显然这与前驱物颗粒成线状的排列方式有关, 但在流动的氧气氛中, 将煅烧温度升到1100 ℃不仅会促进前驱物中未洗净的NH4+ ,NO3- , SO42- 等杂质离子的彻底挥发, 同时也促使前驱物的相变反应完全及生成的Y2O3 完全晶化。因此, 为得到完全晶化并高纯的Y2O3 微粉, 在不使产物粒度过分长大的前提下, 煅烧温度以1100 ℃保温3 h 为佳。
3 结 论
1. SO42- 离子对前驱物颗粒的聚集状态及煅烧产物的颗粒形貌有显著影响, 其作用使前驱物颗粒呈线状排列, 并以雪花放射状结构聚集, 抑制颗粒团聚, 得到前驱物溶胶, 在煅烧过程中起到促进颗粒晶化各向同性发展的作用, 使结晶生成的Y2O3 趋近于球形。
2. 前驱物在煅烧过程中的物相变化依次为:Y2(OH)5(NO3) 0. 86·H2O →α2Y(OH)3 →YOOH →Y2O3 , 物相反应在600 ℃以下已基本完成, 但不完全。
3. 在不使产物粒度过分长大的前提下, 升高煅烧温度, 有助于得到高纯并晶化完全的Y2O3微粉;1100 ℃保温3 h , 得到粒度均匀、一次颗粒平均尺寸为60 nm、形状呈球形的Y2O3微粒。
|